import pandas as pdimport numpy as npimport plotly.express as pximport plotly.graph_objects as goimport astpd.options.mode.chained_assignment =Nonedf_total_flat = pd.read_csv('./www/database/df_flattened.csv')df_total_no_TM = pd.read_csv('./www/database/results_no_TM.csv')list_columns = ['binding_sites', 'area_a2', 'alpha_seeds', 'beta_seeds', 'hydrophobicity']for col in list_columns: df_total_no_TM[col] = df_total_no_TM[col].apply(lambda x: ast.literal_eval(x) if pd.notnull(x) else [])all_classes = df_total_no_TM["main_class"].unique()sub_classes = df_total_no_TM["sub_class_1"].unique()avg_bs_dic = {}protein_per_family_dic = {}for item in sub_classes: all_no_sites = [] protein_count =0for index, row in df_total_no_TM.iterrows():if row["sub_class_1"] == item: all_no_sites.append(int(row["site"])) protein_count+=1iflen(all_no_sites) ==0:print("error")continueelse: avg_bs_dic[item] =sum(all_no_sites)/len(all_no_sites) protein_per_family_dic[item] = protein_countsubclass = [item for item in avg_bs_dic]mainclass = []for subs in subclass:if subs =="Unclassified": mainclass.append(list(set(list(df_total_no_TM[df_total_no_TM['sub_class_1'] == subs]["main_class"])))[0])else: mainclass.append(list(set(list(df_total_no_TM[df_total_no_TM['sub_class_1'] == subs]["main_class"])))[0])Surfaceome = ["Surfaceome"] *22count = [protein_per_family_dic[c] for c in protein_per_family_dic]bs = [avg_bs_dic[b] for b in avg_bs_dic]df = pd.DataFrame(dict(subclass=subclass, mainclass=mainclass, Surfaceome=Surfaceome, count=count, BindingSites=bs))fig = px.sunburst(df, path=['Surfaceome', 'mainclass', 'subclass'], width=800, height=800, values='count', color='BindingSites', color_continuous_scale='RdBu', color_continuous_midpoint=np.average(df['BindingSites'], weights=df['BindingSites']))fig.update_traces(insidetextorientation="radial")fig.update_layout(margin =dict(t=0, l=0, r=0, b=0))# fig.update_traces(textfont=dict(family=['Arial'],size=[0,0,0,0,0]))fig.show()
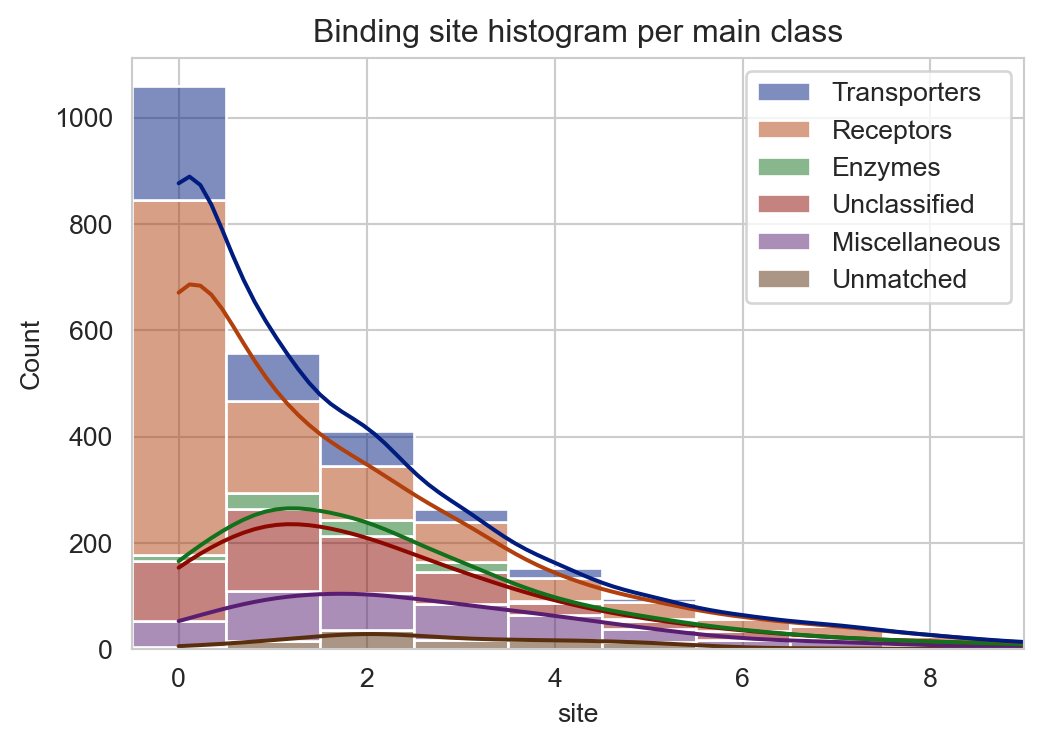
Binding site histogram on main classes
Code
import matplotlib.pyplot as pltimport seaborn as snsplt.figure(figsize=(6, 4))sns.set_style("whitegrid")g = sns.histplot(data=df_total_no_TM, x="site", hue="main_class", multiple="stack", discrete=True, kde=True, palette="dark")g.set(xlim=(-0.5,9))g.legend_.set_title(None)plt.title("Binding site histogram per main class")plt.show(g)
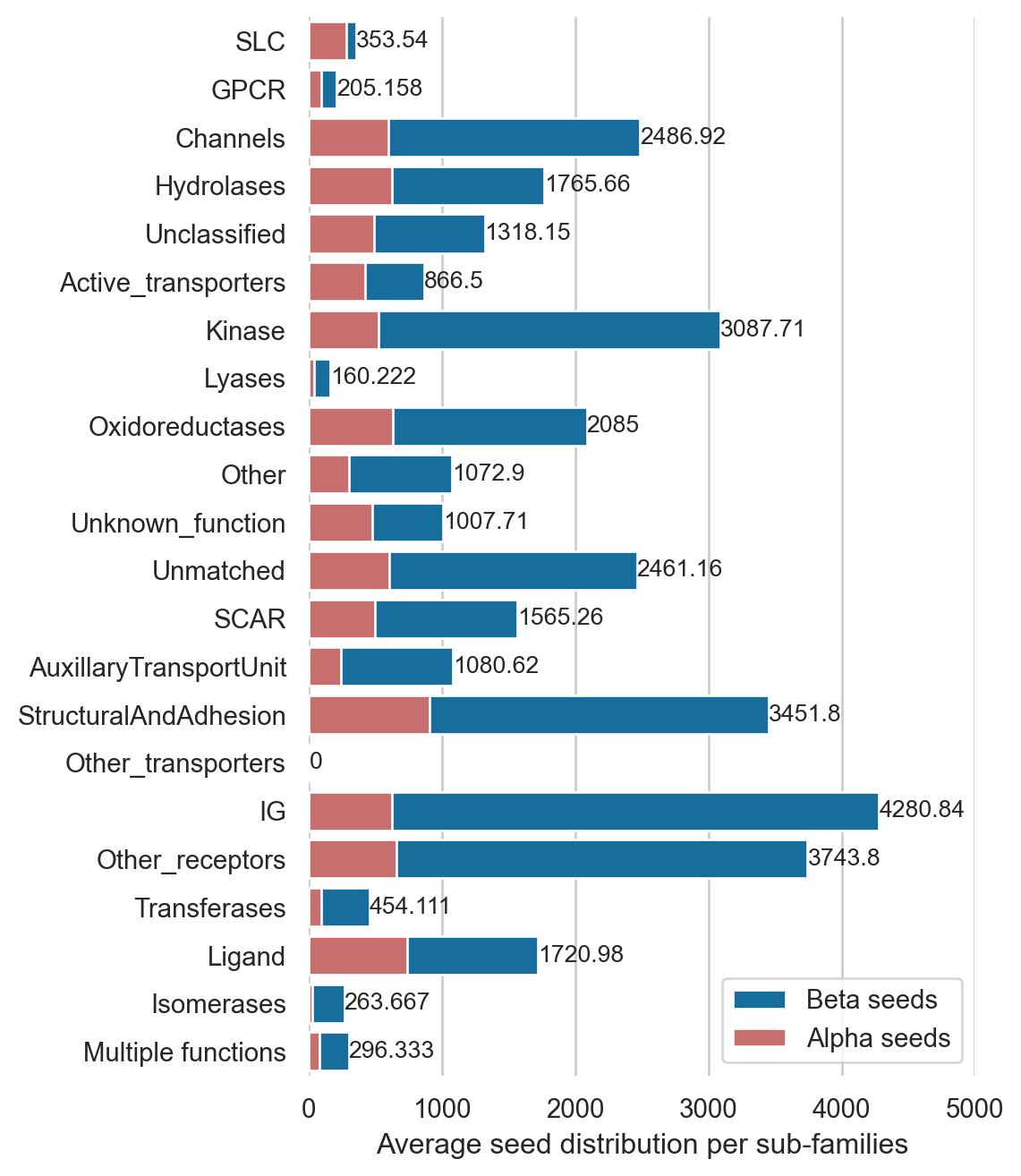
Seed distribution in sub-families
Code
avg_seed_a_dic = {}avg_seed_b_dic = {}for item in sub_classes: no_seeds_a = [] no_seeds_b = []for i, row in df_total_no_TM.iterrows():if row["sub_class_1"] == item: no_seeds_a.append(sum(row["alpha_seeds"])) no_seeds_b.append(sum(row["beta_seeds"])) avg_seed_a_dic[item] =sum(no_seeds_a)/len(no_seeds_a) avg_seed_b_dic[item] =sum(no_seeds_b)/len(no_seeds_b)count = [protein_per_family_dic[c] for c in protein_per_family_dic]bs = [avg_bs_dic[b] for b in avg_bs_dic]seed_a = [avg_seed_a_dic[a] for a in avg_seed_a_dic]seed_b = [avg_seed_b_dic[b] for b in avg_seed_b_dic]df2 = pd.DataFrame(dict(subclass=subclass, mainclass=mainclass, Surfaceome=Surfaceome, count=count, seed_a=seed_a, seed_b=seed_b))import seaborn as snsimport matplotlib.pyplot as pltsns.set_theme(style="whitegrid")f, ax = plt.subplots(figsize=(5, 8))sns.set_color_codes("colorblind")sns.barplot(x="seed_b", y="subclass", data=df2, label="Beta seeds", color="b")sns.set_color_codes("muted")sns.barplot(x="seed_a", y="subclass", data=df2, label="Alpha seeds", color="r")ax.legend(ncol=1, loc="lower right", frameon=True)ax.set(xlim=(0, 5000), ylabel="", xlabel="Average seed distribution per sub-families")ax.bar_label(ax.containers[0], fontsize=10);sns.despine(left=True, bottom=True)
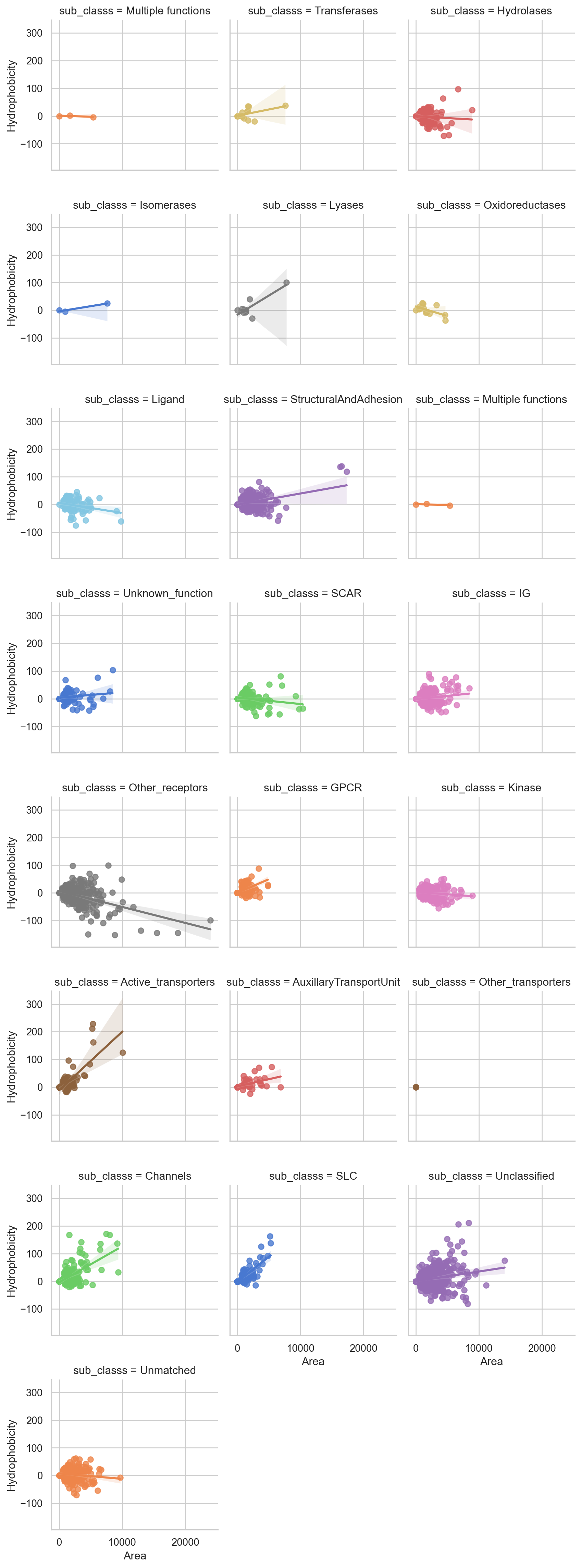
Correlation between Area and Hydrophobicity of the binding sites for Sub-familes