import requestsimport urllib3urllib3.disable_warnings()def fetch_uniprot_data(uniprot_id): url =f"https://rest.uniprot.org/uniprotkb/{uniprot_id}.json" response = requests.get(url, verify=False) # Disable SSL verification response.raise_for_status() # Raise an error for bad status codesreturn response.json()def display_uniprot_data(data): primary_accession = data.get('primaryAccession', 'N/A') protein_name = data.get('proteinDescription', {}).get('recommendedName', {}).get('fullName', {}).get('value', 'N/A') gene_name = data.get('gene', [{'geneName': {'value': 'N/A'}}])[0]['geneName']['value'] organism = data.get('organism', {}).get('scientificName', 'N/A') function_comment =next((comment for comment in data.get('comments', []) if comment['commentType'] =="FUNCTION"), None) function = function_comment['texts'][0]['value'] if function_comment else'N/A'# Printing the dataprint(f"UniProt ID: {primary_accession}")print(f"Protein Name: {protein_name}")print(f"Organism: {organism}")print(f"Function: {function}")# Replace this with the UniProt ID you want to fetchuniprot_id ="Q9BZR6"data = fetch_uniprot_data(uniprot_id)display_uniprot_data(data)
UniProt ID: Q9BZR6
Protein Name: Reticulon-4 receptor
Organism: Homo sapiens
Function: Receptor for RTN4, OMG and MAG (PubMed:12037567, PubMed:12068310, PubMed:12089450, PubMed:12426574, PubMed:12839991, PubMed:16712417, PubMed:18411262, PubMed:19052207). Functions as a receptor for the sialylated gangliosides GT1b and GM1 (PubMed:18411262). Besides, functions as a receptor for chondroitin sulfate proteoglycans (By similarity). Can also bind heparin (By similarity). Intracellular signaling cascades are triggered via the coreceptor NGFR (PubMed:12426574). Signaling mediates activation of Rho and downstream reorganization of the actin cytoskeleton (PubMed:16712417, PubMed:22325200). Mediates axonal growth inhibition (PubMed:12839991, PubMed:19052207, PubMed:28892071). Plays a role in regulating axon regeneration and neuronal plasticity in the adult central nervous system. Plays a role in postnatal brain development. Required for normal axon migration across the brain midline and normal formation of the corpus callosum. Protects motoneurons against apoptosis; protection against apoptosis is probably mediated via interaction with MAG. Acts in conjunction with RTN4 and LINGO1 in regulating neuronal precursor cell motility during cortical development. Like other family members, plays a role in restricting the number dendritic spines and the number of synapses that are formed during brain development (PubMed:22325200)
More information:
AlphaFold model
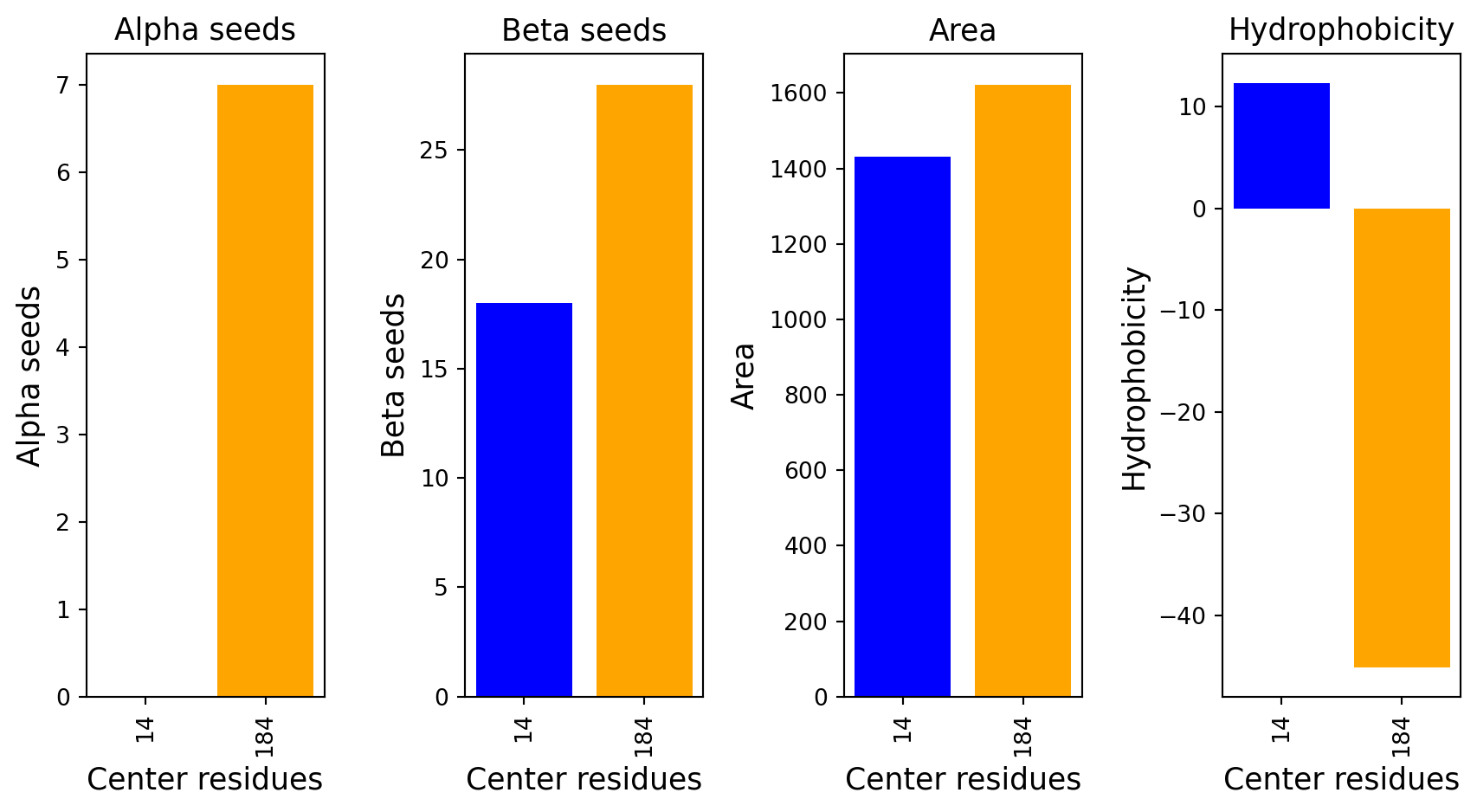
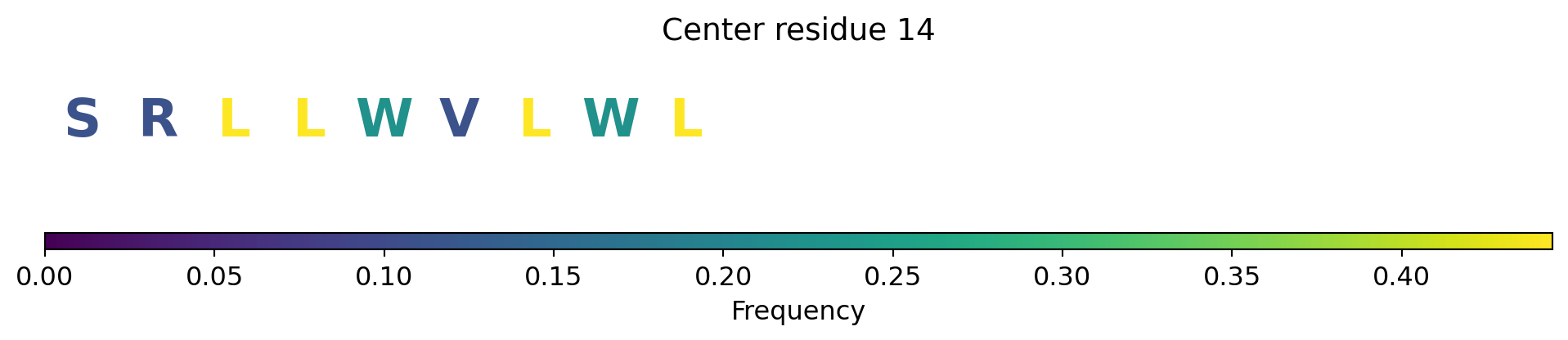
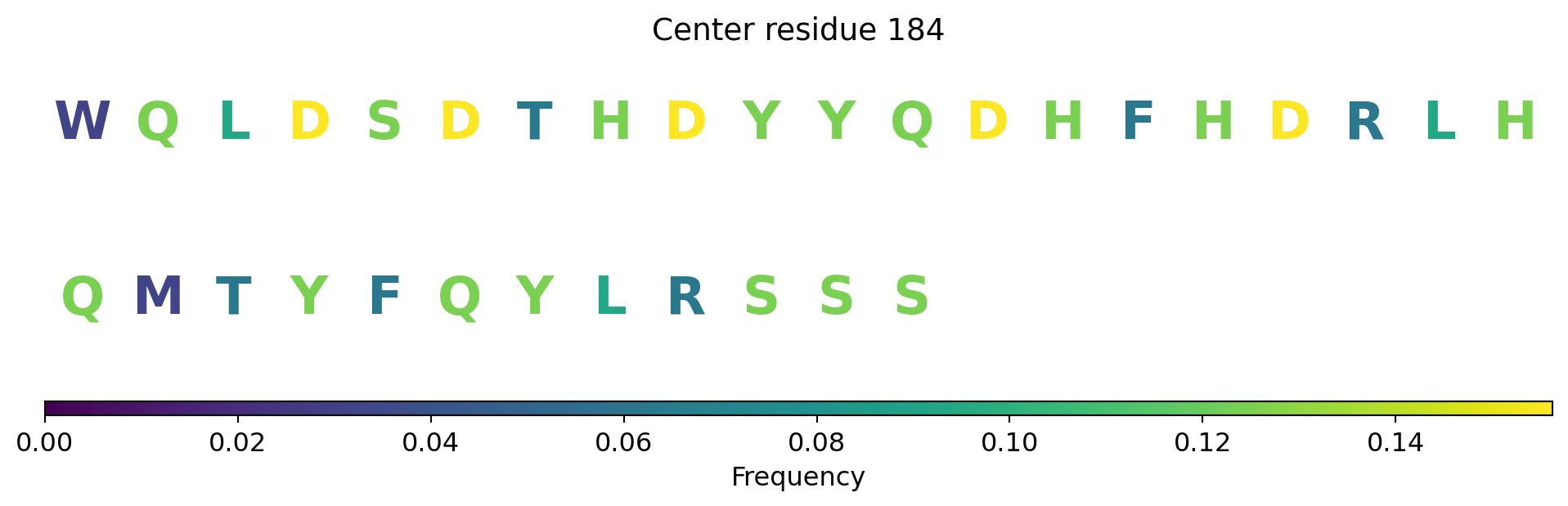
Surface representation - binding sites
The computed point cloud for pLDDT > 0.6. Each atom is sampled on average by 10 points.
To see the predicted binding interfaces, you can choose color theme “uncertainty”.
Go to the “Controls Panel”
Below “Components”, to the right, click on “…”
“Set Coloring” by “Atom Property”, and “Uncertainty/Disorder”