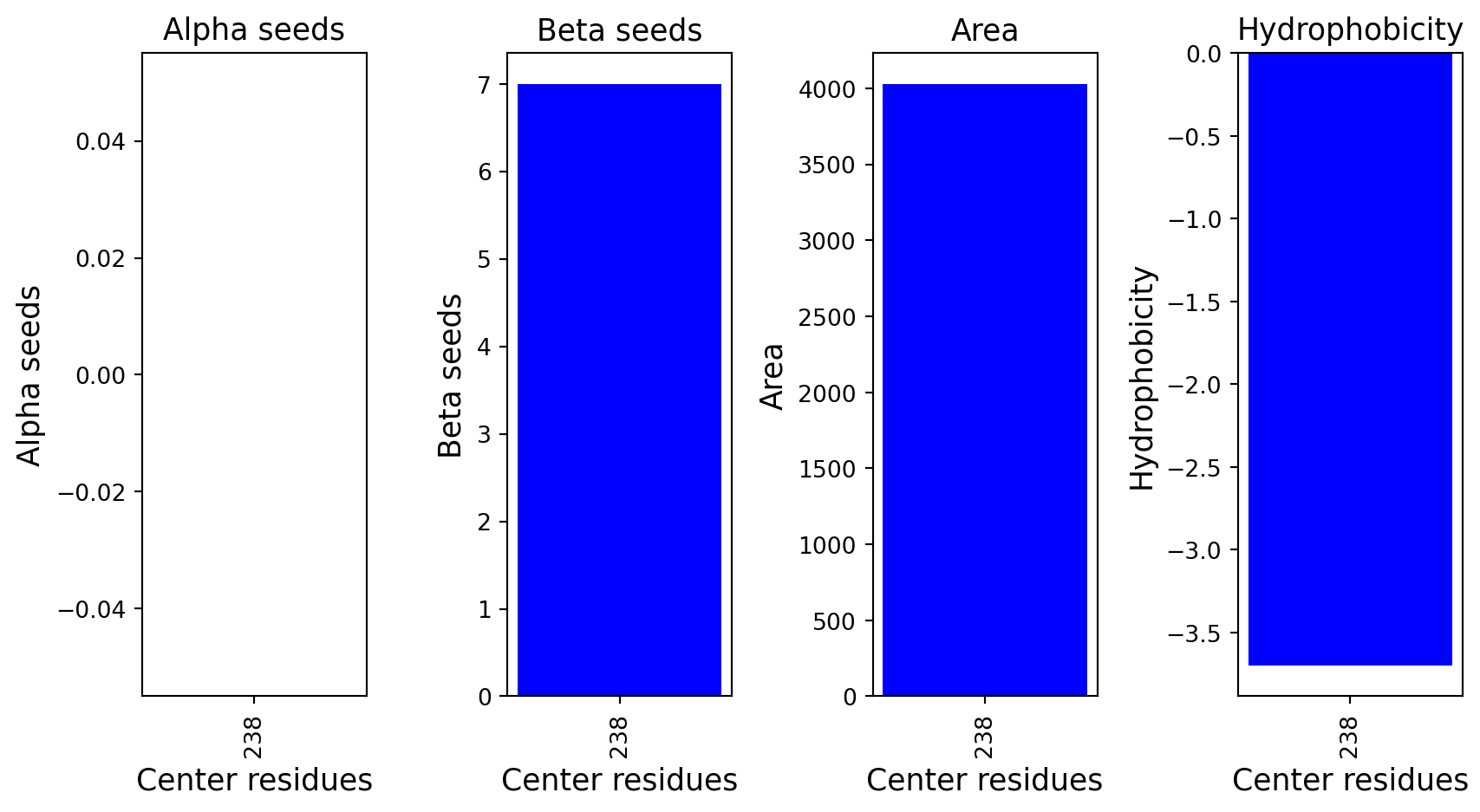
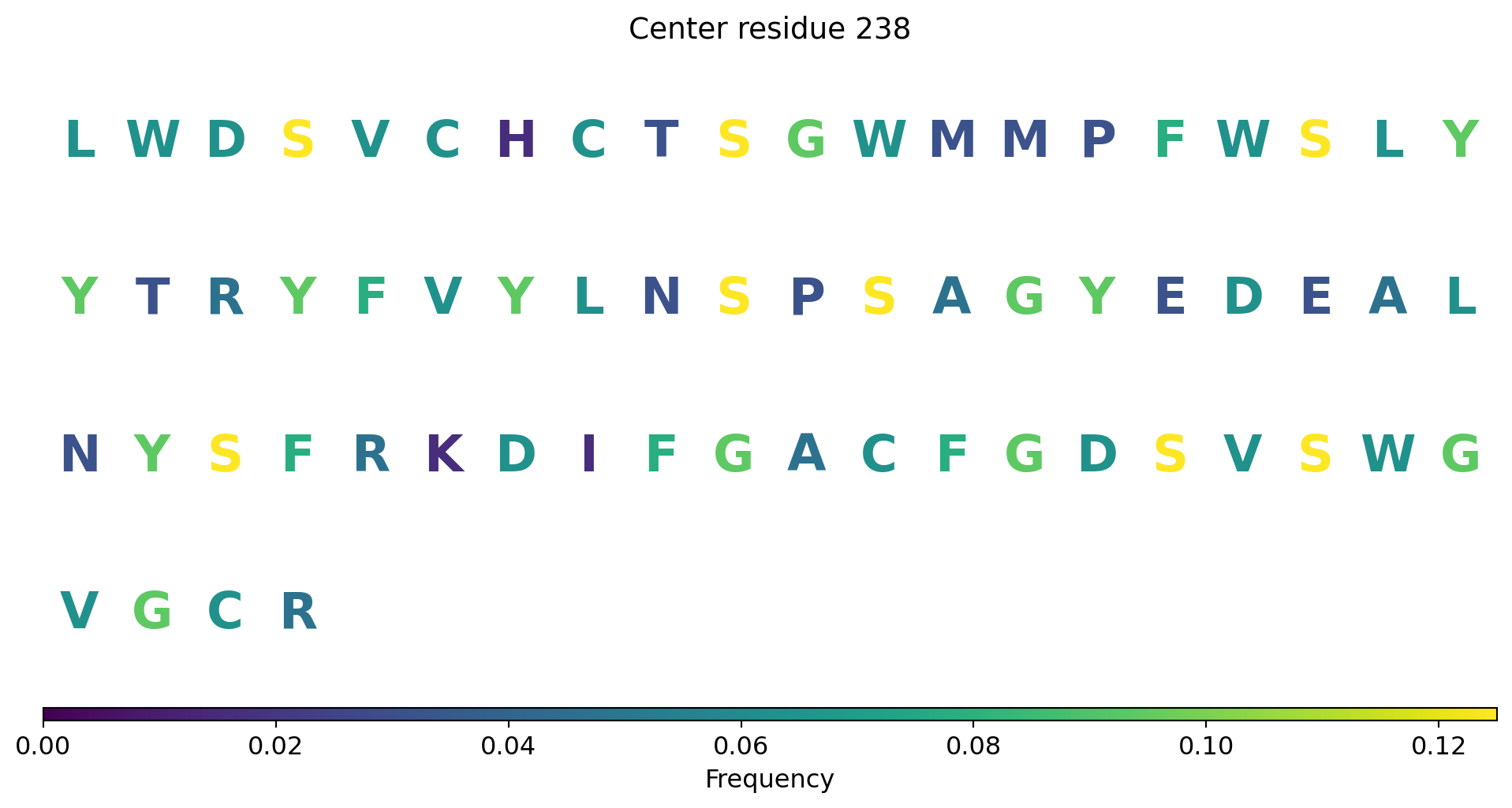
import requestsimport urllib3urllib3.disable_warnings()def fetch_uniprot_data(uniprot_id): url =f"https://rest.uniprot.org/uniprotkb/{uniprot_id}.json" response = requests.get(url, verify=False) # Disable SSL verification response.raise_for_status() # Raise an error for bad status codesreturn response.json()def display_uniprot_data(data): primary_accession = data.get('primaryAccession', 'N/A') protein_name = data.get('proteinDescription', {}).get('recommendedName', {}).get('fullName', {}).get('value', 'N/A') gene_name = data.get('gene', [{'geneName': {'value': 'N/A'}}])[0]['geneName']['value'] organism = data.get('organism', {}).get('scientificName', 'N/A') function_comment =next((comment for comment in data.get('comments', []) if comment['commentType'] =="FUNCTION"), None) function = function_comment['texts'][0]['value'] if function_comment else'N/A'# Printing the dataprint(f"UniProt ID: {primary_accession}")print(f"Protein Name: {protein_name}")print(f"Organism: {organism}")print(f"Function: {function}")# Replace this with the UniProt ID you want to fetchuniprot_id ="Q9Y6M0"data = fetch_uniprot_data(uniprot_id)display_uniprot_data(data)
UniProt ID: Q9Y6M0
Protein Name: Testisin
Organism: Homo sapiens
Function: Could regulate proteolytic events associated with testicular germ cell maturation
More information:
AlphaFold model
Surface representation - binding sites
The computed point cloud for pLDDT > 0.6. Each atom is sampled on average by 10 points.
To see the predicted binding interfaces, you can choose color theme “uncertainty”.
Go to the “Controls Panel”
Below “Components”, to the right, click on “…”
“Set Coloring” by “Atom Property”, and “Uncertainty/Disorder”